Alzheimer’s disease: From Humans to Experimental Models


Our team conducts research on Alzheimer’s disease (AD) and Frontotemporal dementia (FTD) and is committed to identifying better therapeutic targets and finding more effective treatments for these devastating disorders. We formulate and test mechanistic hypotheses on AD and FTD pathogenesis by incorporating cellular and animal models with synergistic studies using postmortem human brain tissue samples. We use highly innovative microscopy, biochemistry, and computational approaches to make discoveries in humans and study mechanisms in experimental models.
We have undertaken innovative computational systems biology approaches to study synaptic dysfunction in AD. We conduct network analyses of proteomics from human brain tissue samples and incorporate orthogonal molecular and cellular traits. Next, protein module-trait correlations are used to guide selection of proteins for functional validation using CRISPR-dCas9 in model systems. Our network approaches enable unbiased identification of proteins functionally involved in disease mechanisms. Our pipeline offers a blueprint to better contextualize omics-based targets with relevant biological mechanisms.
Cognitive Resilience in Alzheimer’s disease
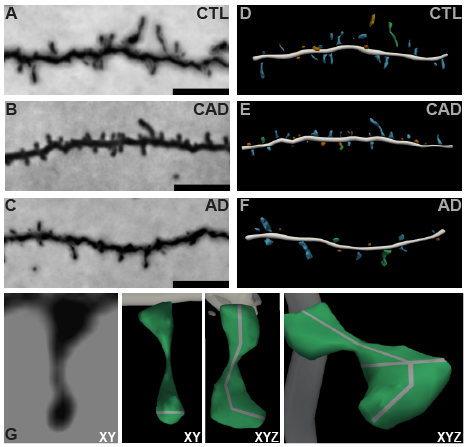
Exciting advances in neuroimaging and other biomarker assays provide the means to detect Alzheimer’s disease (AD) pathology in vivo and yield crucial evidence that the pathological processes of AD initiate years to decades prior to clinical dementia onset. Yet, approximately 30%-50% of individuals who come to autopsy without dementia have high levels of AD pathology. It is hypothesized that such individuals exhibit cognitive resilience that protects against dementia. How cognitively normal older individuals with AD pathology withstand the development of dementia has remained one of the most pivotal, unanswered questions in the field.
A proposed mechanism of cognitive resilience is the hypothesis that the brain is capable of protective structural remodeling in the face of aging and disease. However, studies providing neurobiological evidence of this in patients with AD pathology did not exist. To address this, our team used innovative, highly optimized three-dimensional modeling of dendritic spines to analyze prefrontal cortex synapse populations from controls, cognitively normal individuals with AD pathology, and AD dementia cases. Our analysis shows that dendritic spines undergo unique structural remodeling exclusively in patients with high AD pathology but no cognitive impairment. Our discovery is the first cellular evidence for cognitive resilience in humans with AD pathology. Current projects in the lab aim to provide deeper insight on this natural, neurobiological mechanism that may explain how individuals with no cognitive impairment withstand AD pathology. Our novel approaches will enable more focused and directed mechanistic and therapeutic studies of cognitive resilience against AD dementia in both humans and experimental model systems.
Intracellular Signaling and Structural Plasticity
Communication among neurons is mediated through synaptic connections between axons and dendrites, and most excitatory synapses occur on actin-rich protrusions along dendrites called dendritic spines. Dendritic spines are structurally dynamic, and synapse strength is closely correlated with spine morphology. Abnormalities in the size, shape, and number of dendritic spines are prevalent in neurologic diseases, including autism spectrum disorders, schizophrenia, and Alzheimer’s disease.
Rho GTPases are extensively studied regulators of actin dynamics and heavily influence dendritic morphology and synaptic plasticity. Rho GTPases can signal through two pathways: (1) activation of Rho-associated protein kinases (ROCK)s > phosphorylation of LIM kinases > phosphorylation of cofilin > actin remodeling, and/or (2) ROCKs > phosphorylation of myosin light chan phosphatase > myosin light chain > actin remodeling. However, it is currently a mystery how these individual signaling cascades alter dendritic morphology in neurons. This remains a critical barrier to neurologic disease research. To overcome this, our team is working on combinatorial genetic and pharmacologic strategies to understand how these pathways signal on actin and may convey therapeutic benefit for neurologic disorders that feature dendritic spine loss or aberrant structural plasticity.

